
From Blobs to Atoms: The Long Journey of Cryo-Electron Microscopy
How physics, chemistry, and computation came together to bring biology into focus.

In 2013, cryo-electron microscopy entered what is called the “resolution revolution,” when it became possible to image large biomolecules at near-atomic resolution. In 2017, Richard Henderson, Joachim Frank, and Jacques Dubochet shared the Nobel Prize in Chemistry for developing cryo-electron microscopy (cryo-EM). But the electron microscope itself was invented in 1931, and Ernst Ruska, its creator, won the Nobel Prize for its development in 1986. So then why did a microscope that was invented in 1931 win another Nobel Prize almost ninety years later? What were the most recent breakthroughs?
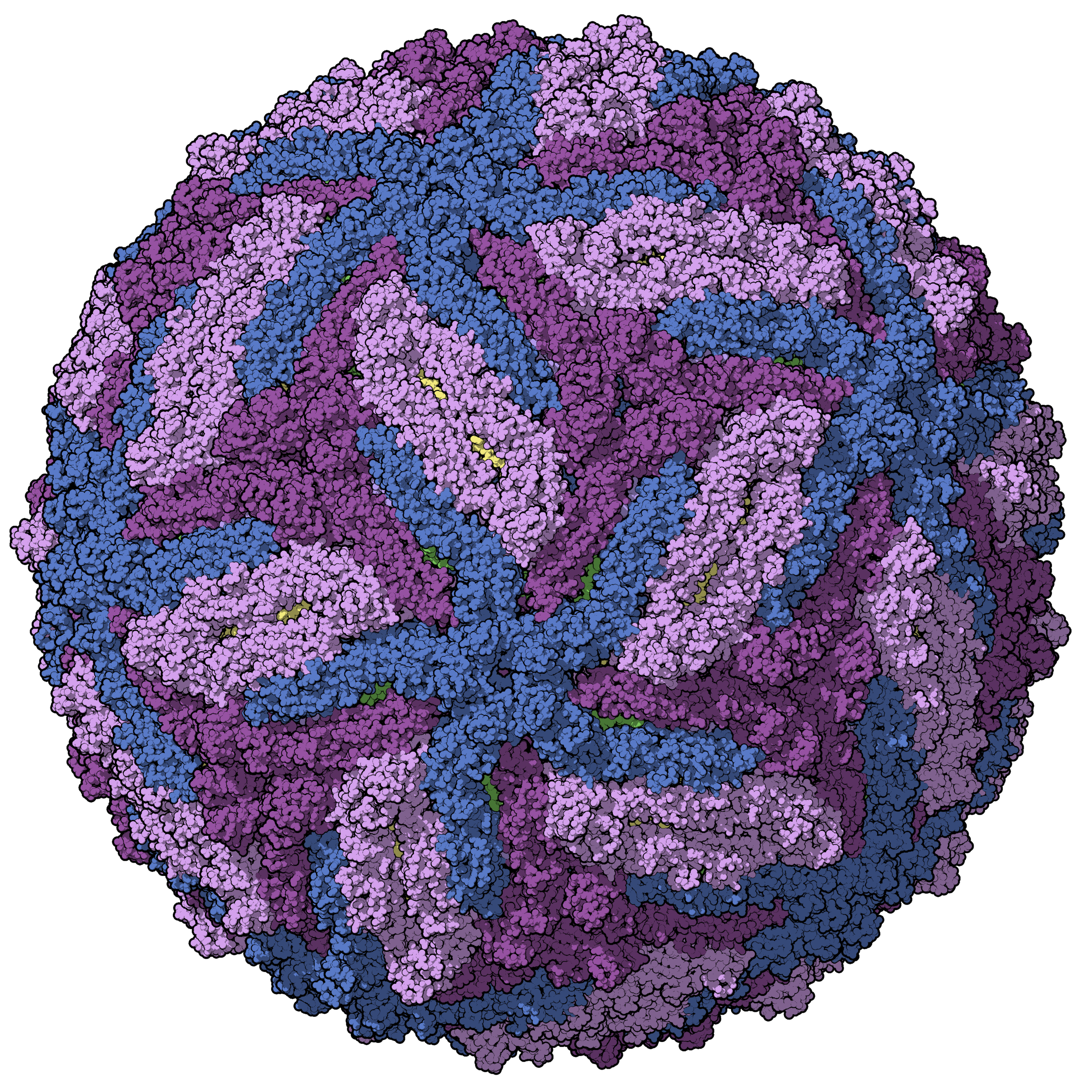
Ever since the invention of the microscope, scientists have wanted to expand the depths of what they could see into plant and animal life. Biology spans a vast spatial spectrum from angstroms to centimeters. On the large end we have whole organisms, which themselves vary in size from single-celled bacteria to large mammals. At the small end we find proteins and individual amino acids that build up these protein chains. In between we have everything from viruses to sub-cellular organelles. From Antonie van Leeuwenhoek and Santiago Ramon y Cajal to Ed Boyden and Richard Henderson, we have traveled the spatial scale of millimeters, micrometers, nanometers, angstroms, and are now closing in on the sub-angstrom world.
In my previous piece, Zooming into biological structure, I wrote about how scientists have built a mosaic of microscopy techniques to study biological structure. I also recently published a piece with Asimov Press, Making the Electron Microscope, which talks about the early history of the electron microscope and a detailed description of how it works. This current essay, which is meant to be a standalone piece, focuses on the more recent history of electron microscopy. I outline how an amalgamation of sample preparation techniques, computational advances, and electron detector technology all came together to revolutionize structural biology, yet again.
Today, cryo-EM allows us to resolve biological structures at the scale of individual atoms. In 2020, a group of scientists at the MRC Laboratory of Molecular Biology in Cambridge, UK, resolved the structure of a neuronal membrane protein at atomic resolution. In 2024, a group at the University of Leeds determined the structure of tau protein and amyloid-β within postmortem tissue from Alzheimer’s disease brains to near-atomic resolution. Efforts are now underway to use these three-dimensional structures to design drugs that target specific domains within the structure.
Progress in science often depends not on one breakthrough, but on the slow alignment of many. In the case of electron microscopy, the revolution came only after decades of parallel advances in chemistry, physics, and computation finally converged.
For the purpose of this essay, I will only briefly describe how the electron microscope works. For full detail I refer you to the Asimov Press piece. At its core, the electron microscope works much like a light microscope, but instead of photons it uses a beam of electrons to probe the sample. At the top of its tall metal column sits the electron gun that emits electrons accelerated to nearly the speed of light. Magnetic lenses made of coiled wire focus and steer this beam, just as glass lenses bend light. When the electrons pass through a thin sample, they scatter according to the arrangement of atoms inside, carrying with them information about the sample’s structure. These scattered electrons are magnified through a series of lenses and projected onto a detector, forming an image. Because electron wavelengths are thousands of times shorter than visible light, the microscope can resolve features far smaller than anything seen with optical microscopy, down to individual atoms under the right conditions.
If you freeze water fast enough, it will turn glass-like
While electron microscopy promised theoretical resolutions at the scale of atomic bonds, in practice this proved extremely difficult to achieve. For decades, it remained a niche tool with several major limitations. One of the biggest challenges was the water present in biological samples, a problem that’s hard to avoid, since most of biology is made of water. As I described in my Asimov piece:
“For instance, from the start, electron microscopy for biology faced a water problem. Because the microscope operates under high vacuum, liquid water evaporates instantly, leaving delicate biological samples collapsed or distorted. To avoid this, aqueous samples had to be dried, fixed, or stained, which produced recognizable images but with obvious artifacts, such as shrunken cells, ruptured membranes, and structural distortions that no longer reflected the living state. Through the 1940s and 1950s, embedding samples in resins and the use of ultra-thin sectioning made cellular ultrastructure visible, while freeze-drying and early cryogenic sectioning offered partial preservation of hydrated material, though the results were still plagued by distortion.”

Another major limitation was radiation damage. The high-energy electron beam that revealed such fine detail also destroyed the very sample it was imaging, burning through delicate biological material within seconds. Scientists realized that cooling the sample below freezing reduced molecular motion, which slowed heat buildup and therefore limited radiation damage. The problem, however, was water itself: when it freezes, it expands and forms crystalline ice, which distorts biological structures.
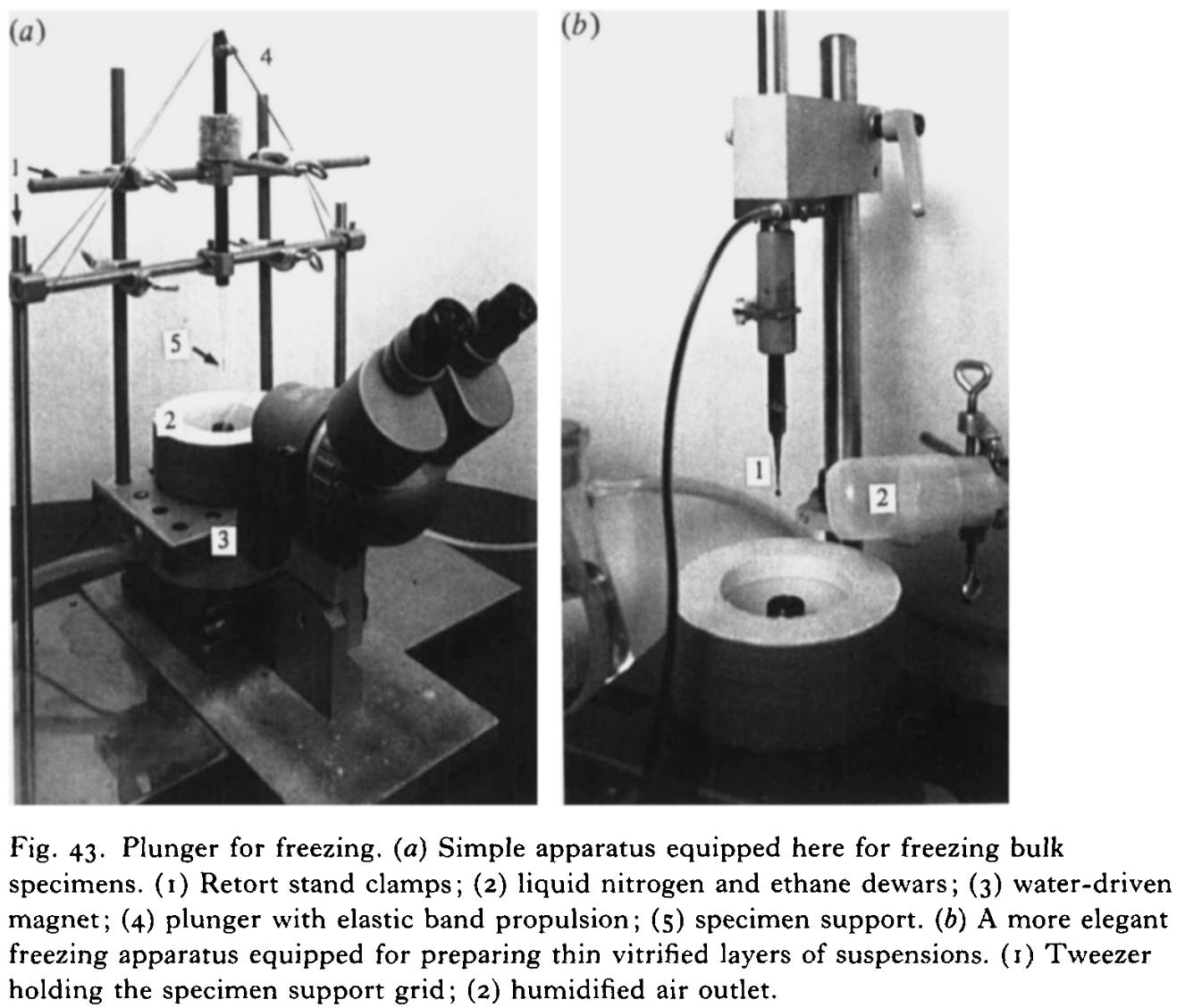
The breakthrough came with the realization that if you freeze water fast enough, the molecules don’t have time to form a crystal lattice. Instead, they solidify into an amorphous, glass-like state in a process called vitrification. This is exactly what Jacques Dubochet and his colleagues achieved in the 1980s at the European Molecular Biology Laboratory. They demonstrated that water could be rapidly cooled into vitreous ice, thereby preserving biological samples in a lifelike, hydrated form, without the destructive crystallization of ice.
Today, samples are typically vitrified by plunging them into liquid ethane chilled by liquid nitrogen to around -183 °C. In an instant, the thin film of water around the molecules solidifies into clear, glass-like ice. This technique not only reduces radiation damage but also preserves biomolecules almost exactly as they exist in life. They are frozen in motion, yet structurally intact.
If you average enough particles, you get three-dimensional models
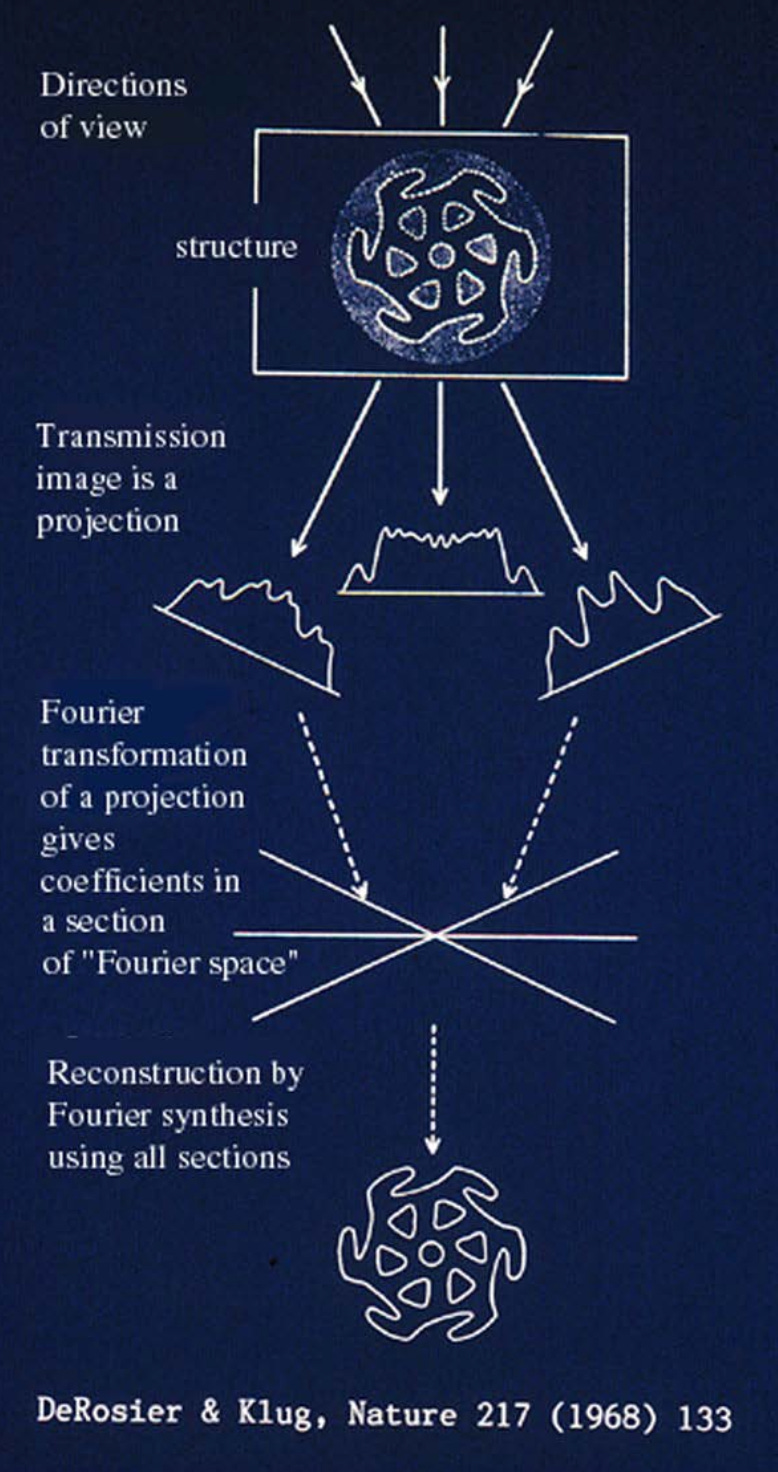
Another major bottleneck for electron microscopy was the lack of three-dimensional information. For that, X-ray crystallography remained the gold standard. Since the 1930s, X-ray crystallography had enabled atomic-resolution structures of biological molecules, but it came with a key requirement: the ability to produce high-quality crystals. These crystals diffracted X-rays at specific angles to reveal the spacing of atoms within the crystal. However, many large macromolecules, such as viruses, ribosomes, or membrane proteins, could not be crystallized, leaving major regions of biology structurally unresolved.
In contrast, electron microscopy, despite its much higher theoretical resolution, produced only two-dimensional projection images of samples. For decades, electron microscopy was primarily a visualization tool for large complexes and cellular structures, but reconstructing accurate 3D models from these projections remained a formidable challenge. Without the regular, repeating patterns that crystals provided, early scientists lacked the computational methods to extract reliable 3D information.
That changed when Joachim Frank and his colleagues developed statistical and computational techniques to solve this problem. As I wrote in my Asimov piece:
“In parallel, computational techniques were improving. Beginning in the 1970s, Joachim Frank developed statistical methods for aligning and averaging thousands of noisy electron micrographs of individual macromolecules. This “single-particle analysis” transformed faint, low-contrast images into coherent 3D reconstructions. When combined with Dubochet’s vitrification method, the two advances gave rise to single-particle cryo-electron microscopy: Molecules suspended in vitreous ice could be imaged in random orientations and computationally combined into detailed three-dimensional structures.”
To unpack this a bit more: a single vitrified sample of purified protein may contain millions of identical macromolecules, such as ribosomes, each embedded in a thin layer of vitreous ice. Before freezing, these molecules move freely in solution. Upon vitrification, they are trapped in place, ideally in random orientations1, producing a field of particles viewed from many angles. Frank’s methods used statistical alignment and averaging to identify the orientation of each particle and combine these thousands of 2D projections into a coherent 3D reconstruction.
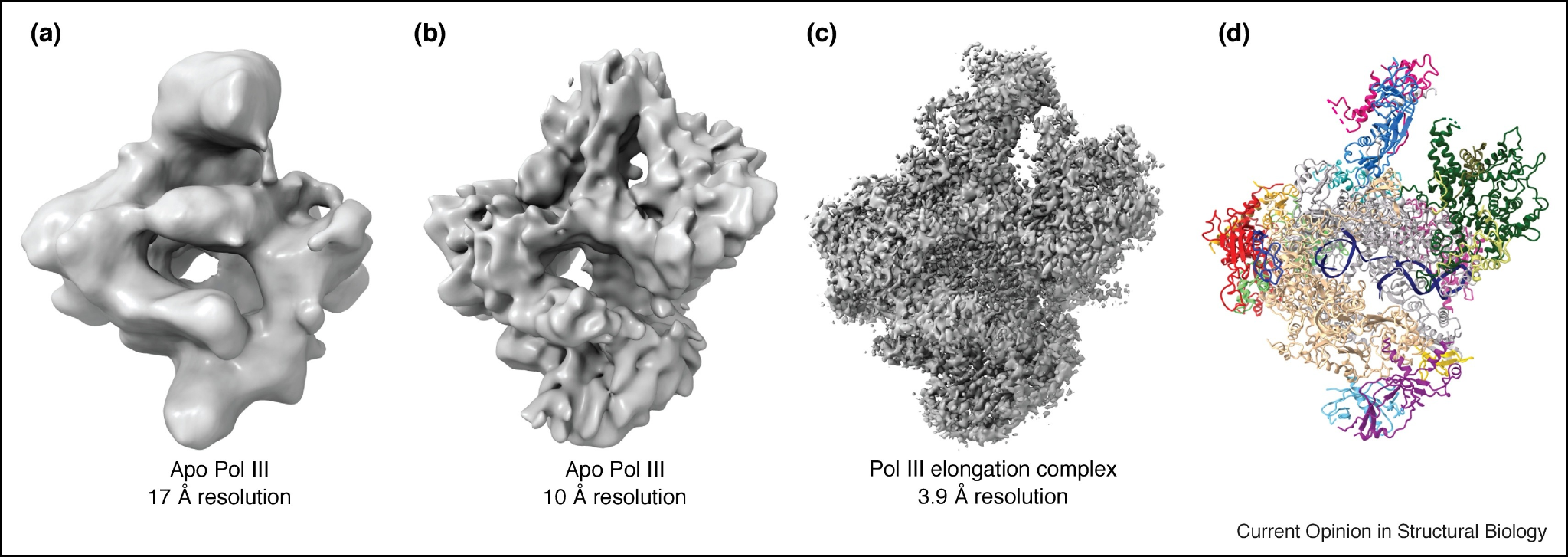
In the 1970s and 1980s, these images were recorded on photographic film that was later digitized for analysis. By the 1990s, CCD cameras began to replace film, streamlining data collection, though they still offered limited resolution. With these early tools, single-particle reconstructions typically reached resolutions of about 10 to 20 angstroms, enough to reveal the overall shape of large complexes like ribosomes, but not the positions of individual amino acid side chains.
While vitrification and computational methods had laid the foundation, the achievable resolution was still constrained by the quality of the recorded images. To break through that final barrier, a third component needed to fall into place.
If you detect electrons directly, you get a revolution
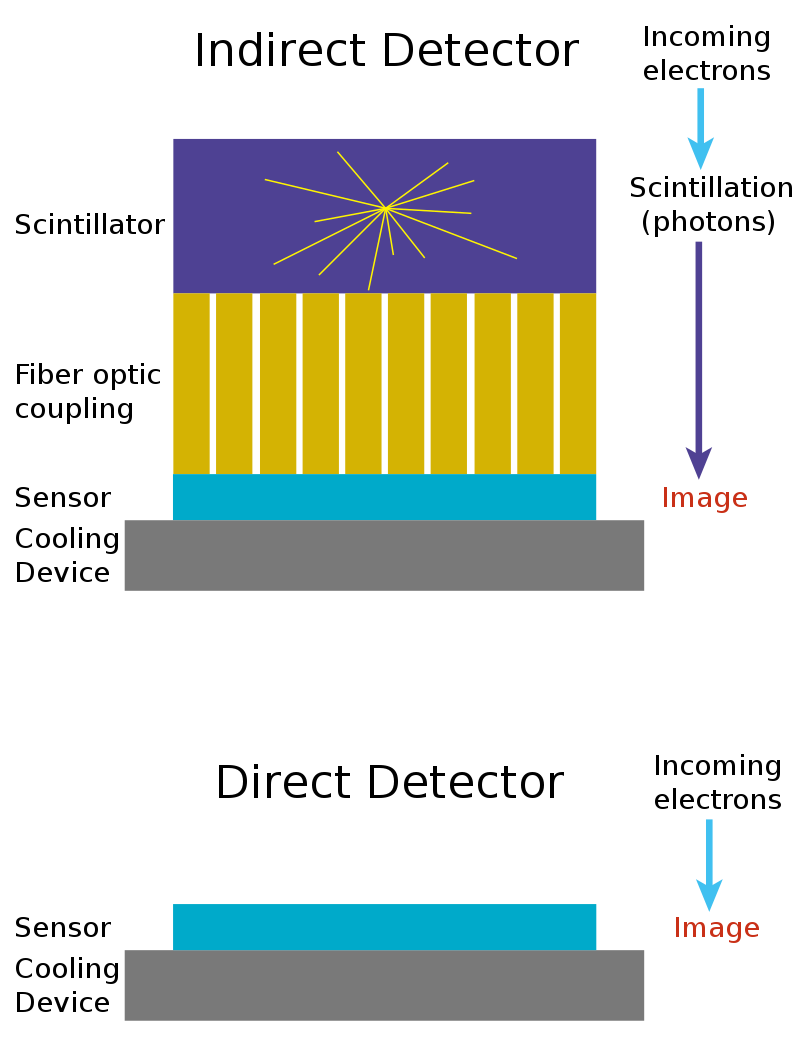
The stagnation in resolution was now due to the efficiency of the camera detectors. CCD and CMOS sensors were originally designed to detect light, not electrons. Light, in the form of photons, is converted into electrical charge and read by the sensor. Electrons, however, are not light. To overcome this limitation, scientists added a scintillator in front of the detector, which is a layer that converts incoming electrons into photons, which are then detected by the camera. But when electrons strike the scintillator, the emitted photons scatter in all directions, blurring the image and reducing resolution.
While digital cameras allowed faster feedback and easier image analysis, their scattering effect meant that photographic film still outperformed digital acquisition in terms of resolution. By the early 2000s, it was clear to nearly everyone in the field that this indirect detection method was fundamentally inefficient. The next leap in resolution would require detecting electrons directly.
Traditional CCD and CMOS cameras weren’t built for this task. They were designed for photons, which interact only with the surface of a sensor. High-energy electrons, by contrast, penetrate deeply into the silicon, creating trails of electrical charge that spread out through the material. In these older chips, the regions that collect the signal and the regions that read it out overlapped. Because the charge was generated deep inside the silicon, much of it was lost before reaching the collection layer. Even the charge that did make it to the surface often spread across several neighboring pixels, a phenomenon known as charge spreading, which blurred fine details and limited resolution.
{kind=link}
The solution came with a new detector architecture called monolithic active pixel sensors (MAPS). These sensors separated the layer where the signal is generated from the layer that reads it out. They used a thin, lightly doped epitaxial layer2 near the surface of the silicon to collect charge efficiently without interference from the deeper circuitry. This design sharply reduced signal loss and charge spreading, allowing each electron to be detected cleanly and precisely. MAPS were first developed at the Rutherford Appleton Laboratory in the UK for particle physics experiments and published in 2000.
Around this time, the electron microscopy community began experimenting with a variety of new detectors to push performance further. In 2002, Richard Henderson and colleagues demonstrated the use of a hybrid pixel detector for electron microscopy, which separated the sensing and electronics layers but suffered from large pixel sizes that limited resolution. Then, in 2005, at least two independent groups (including Henderson’s) published promising results using MAPS for electron microscopy. These sensors could detect single electrons with excellent signal-to-noise ratios, but they still required engineering refinements before being integrated into commercial instruments.
By 2010, MAPS-based direct electron detectors (DEDs) had been optimized for stable, sustained electron microscopy use and incorporated into commercial microscopes. What made these detectors revolutionary was both their sensitivity to electrons and their speed. Instead of recording a single image on film, scientists could record whole movies, capturing many frames per second. This not only allowed more averaging to get sharper images, it also allowed them to correct for beam-induced motion, which are tiny shifts and vibrations caused by the electron beam that blur every image.
Just three years later, in 2013, the first generation of high-performance DEDs became widely available. For the first time, cryo-EM could capture movies of electrons hitting the detector and reconstruct macromolecules at near-atomic resolution. What had been a decades-long march of incremental improvements suddenly became a resolution revolution, reshaping structural biology and opening a new era of molecular discovery.
A new age in electron microscopy
Imagine a world where everything around you is just slightly out of focus. You can see the trees, but the leaves blur together: shapes without edges, outlines without detail. If you wear glasses, you know the moment that changes, the instant you put them on and the world snaps into focus. I still remember the first thing I said when I tried mine for the first time: “Oh my god, I can see every single leaf on that tree!”
That’s what the resolution revolution of the early 2010s felt like for electron microscopy. After decades of “blobology,” where proteins appeared only as vague, featureless shapes, they could suddenly see the contours of individual amino acids in exquisite detail. But these scientists weren’t looking at trees, they were looking at the machinery of life itself. And I imagine many of them had the same reaction: “Oh my god, I can see every single amino acid in that protein!”
This leap in clarity didn’t come from a single breakthrough. What I love about the story of cryo-EM is that it needed multiple independent lines of long-arc research directions to mature and align. The whole is truly greater than the sum of its parts. Dubochet’s vitrification preserved biological molecules in their native, hydrated state, allowing them to be imaged without distortions. The direct electron detectors were advances on camera technology that captured those fine details with minimal blurring. And Frank’s computational methods leveraged statistics to build clear images from noisy, low-contrast individual micrographs.

Cryo-EM needed all three because its power rests on a delicate sequence: the molecule must first be preserved in a lifelike state, then imaged faithfully, and finally reconstructed computationally. If any one of these steps fails, such as the ice too thick, the camera too inefficient, or the algorithms too crude, the final structure collapses into blur. In that sense, the resolution revolution wasn’t a single eureka moment but the culmination of decades of parallel progress in chemistry, computation, and instrumentation.
A fair question to ask is: did it really need to take that long to achieve atomic resolution? Frank was already developing single-particle averaging methods in the 1970s, and Dubochet’s vitrification techniques appeared in the early 1980s. Yet the so-called “resolution revolution” only arrived more than thirty years later.
Part of the answer lies in the detectors. In the 1980s and 1990s, digital imaging itself was still in its infancy. CCD and CMOS cameras were only beginning to replace photographic film, and while they offered convenience, they couldn’t yet match film’s resolution or sensitivity. The problem wasn’t in the lack of ideas, virtually everyone in the field knew that new detectors were needed. The problem was in the actual development of the hardware. Electron detectors had to evolve dramatically before they could capture images with the precision needed for atomic resolution.
But this delay was also not just technological developments, it was translational too. Even once new detectors like MAPS were demonstrated in physics labs in the early 2000s, it took another decade of engineering and iteration before they could be made robust, affordable, and stable enough for daily use in commercial electron microscopes. Moving a technology from a prototype on a lab bench to an instrument that thousands of scientists can rely on is slow, exacting work. It takes the persistence, and often invisible labor of scientists like Henderson to work alongside engineers to bridge the gap between discovery and adoption.
Looking ahead
This does not conclude my stories on electron microscopy!
For decades, most structures deposited in the PDB came from X-ray crystallography. That balance is now shifting. Over the past ten years, the number of structures solved by cryo-EM has risen sharply, while X-ray crystallography is starting to plateau. If current trends continue, 2025 may mark a symbolic crossover point, where cryo-EM and X-ray crystallography reach equal footing in the number of structures deposited this year in the PDB, and from there, cryo-EM might begin to dominate.
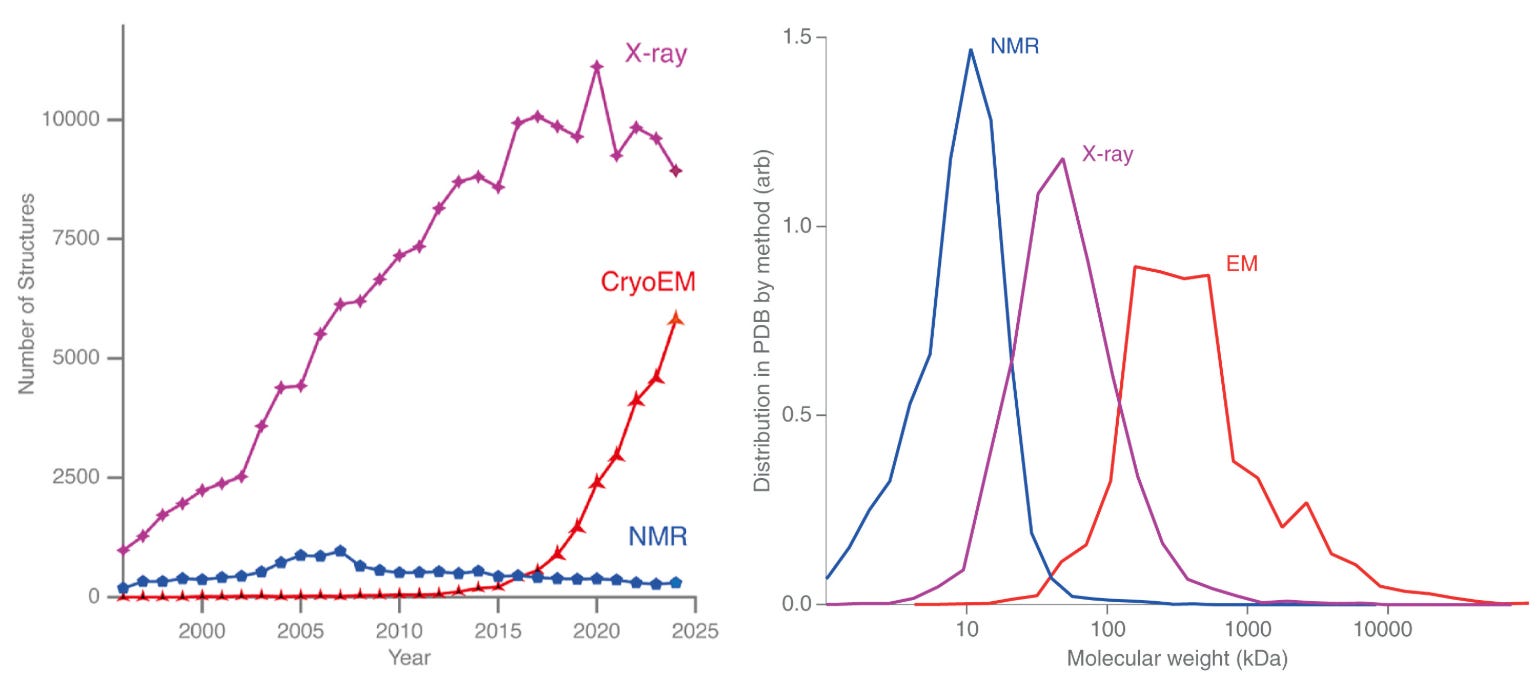
This shift has been driven in large part by the democratization of cryo-EM. The growth of national and regional centers that make cutting-edge microscopes and sample preparation accessible to scientists who might never have had direct access before. These shared facilities have lowered barriers to enter the field of cryo-EM and opened the door for an entirely new generation of structural biologists.
However, sample preparation remains a big, manual, bottleneck. Sample preparation, which requires plunge freezing the specimen into liquid ethane and then manually transferring it to liquid nitrogen, is a skill to be mastered. While it is certainly more automated today than when Dubochet first developed the method, it continues to be as much an art as a science. Producing the perfect thin, vitrified ice, free of contamination or crystal formation, takes remarkable skill.
If we want a future where AI systems will unlock new scientific discoveries, we need large, comprehensive, and consistent datasets for it to learn from. A bigger question may be: what would it take to make the cryo-EM pipeline so consistent and reproducible that an AI could navigate it end-to-end? This question deserves a post of its own.
And we haven’t yet touched the next phase of electron microscopy: cryo-electron tomography (cryo-ET), where scientists tilt samples inside the microscope to reconstruct entire cells and tissues in 3D, not just individual molecules. Cryo-ET is extending the reach of structural biology from purified proteins to the molecular architecture of life itself, captured in its native environment. With it, scientists are beginning to visualize how viruses assemble and mature, how misfolded proteins trigger neurodegeneration, and how organelles organize the crowded interior of the cell. It hasn’t yet reached atomic resolution, but it’s only a matter of time, and sustained effort by motivated individuals, before it does.

Ultimately, the structure of biology is valuable only insofar as it reveals function, and function rarely resides in isolated molecules. Cryo-EM and cryo-ET are bringing us closer to seeing how those molecules work together, interact, and organize into the living systems that make up our world.
Sometimes molecules tend to stick to the surface of the ice in a certain way, so they all end up facing the same direction. This makes it hard to get views of the molecule from all angles, which can blur or distort the 3D reconstruction.
Which just means that it is a top layer.