
How do we diagnose Alzheimer’s Disease?
And why an early diagnosis is important, but still lacking.

Back in May, I saw a post from Bill Gates about the FDA’s approval of the first blood-based test for Alzheimer’s disease. It caught my attention, but it also left me with more questions than answers. What exactly was this blood test measuring? How do you even diagnose Alzheimer’s in the first place? And could a simple blood draw really tell us something useful about such a complicated brain disease?
Since then, I have been trying to learn more about the Alzheimer’s diagnostic space. This essay is part of that process. I am not an expert on Alzheimer’s research. I have worked for years at the intersection of neuroscience and microscopy, but not directly on Alzheimer’s. So this is me, sorting through what I’ve read, what I think I understand, and what still confuses me.

Alzheimer’s affects roughly 40 million people worldwide, and there is no cure. With an aging population, it has never been more crucial to fight Alzheimer’s disease. Several drugs have been tested, but they are modest at best in slowing cognitive decline. One possible reason is that by the time clinical symptoms appear, which is when most patients are diagnosed, the disease biology is already far advanced. Detecting it earlier could give therapies a much better chance of making a difference.
An early and accurate diagnosis matters for more than just treatment. It shapes care plans, determines eligibility for clinical trials (often where the most promising therapies are available), helps rule out reversible causes of cognitive decline, and gives patients and families clarity as they navigate a frightening and confusing situation.
The problem is that our current diagnostic tools are either invasive (like spinal taps) or expensive (like PET scans). In practice, they are usually performed only after symptoms are obvious. That is why blood-based biomarkers are so appealing: they promise a cheap, simple test that could be used earlier and more broadly. And that is why the FDA’s approval in May 2025 felt like such big news.
But as I started reading more, I realized that flashy headlines can be misleading. So I wanted to understand: what exactly makes Alzheimer’s so hard to treat? How is it currently diagnosed? Why are existing tests inaccessible at early stages? And does this new blood test really bring us closer to early detection?
Alzheimer’s begins ~15 years before clinical symptoms appear
Pathologically, Alzheimer’s disease is defined by the accumulation of amyloid-beta plaques outside neurons and tau neurofibrillary tangles inside neurons, leading to widespread neurodegeneration (more on these below). Clinically, it presents as a progressive decline in memory, thinking, and daily functioning, beginning subtly and advancing to severe dementia.
While there are many hypotheses for what causes Alzheimer’s disease, the truth is we still don’t fully know. It’s not for lack of trying. We have tried. A lot. But the biology of how Alzheimer’s begins and unfolds is extremely complex, with feedback loops that play out over decades. With every paper I read, the disease only seems more complex, not less. What we do have a better handle on is how the disease progresses biologically, even if the exact causal links remain unclear.
One of the earliest changes we see is the buildup of a small protein fragment called amyloid beta (Aβ). In particular, the Aβ42 variant tends to deposit in the spaces outside neurons, forming plaques. Aβ is a byproduct of normal healthy processing, when the amyloid precursor protein (APP) is cut. APP is normally produced in healthy brains and may play roles in antimicrobial defense, neuronal signaling, and even plugging leaks in the blood-brain barrier after injury. The problem arises when Aβ is overproduced or not cleared efficiently. In that case, it begins to accumulate into plaques that gradually spread. Over the course of 15 to 20 years, this pathology can extend across much of the brain.
Even so, amyloid on its own turns out not to be a good predictor of cognitive decline. Some people have widespread Aβ plaques and show only subtle, even undetectable, deficits on neurological exams. What does line up more closely with neurodegeneration and symptoms is tau.
Tau is another naturally occurring protein in the brain, responsible for stabilizing the microtubules that form the cell’s internal scaffolding and transport system. But tau can go wrong. When it becomes abnormally folded, it can “seed” nearby tau proteins to misfold as well, creating tangles. These tangles disrupt neurons and trigger neurodegeneration. Memory loss is usually the first noticeable clinical sign of Alzheimer’s because tau pathology begins in the brain’s memory centers. Once it starts, tau can spread rapidly along neural circuits, driving degeneration wherever it goes.
Crucially, once tau pathology is underway, it no longer seems to depend on amyloid. This has potentially big implications for treatment: if you clear amyloid after tau tangles have already formed, you may not stop the damage. Tau pathology, neurodegeneration, and cognitive decline tend to track together in both time and space. That suggests two possible therapeutic windows. One is to target tau directly before neurodegeneration has set in, though this window may be very narrow. The other is to intervene earlier, by clearing amyloid before tau pathology begins.
Current treatments have been modest at best
The first drug to reach patients was aducanumab (Aduhelm). In June 2021, the FDA granted it accelerated approval on the basis that it cleared amyloid plaques from the brain. But the clinical data were inconsistent: one phase 3 trial suggested a modest slowing of decline, while a second found no benefit. The approval was deeply controversial, with several members of the FDA advisory panel voting against it or resigning in protest. By January 2024, Biogen withdrew the drug from the U.S. market.
The next drug to hit the market was lecanemab (Leqembi). It earned accelerated approval in January 2023 and full traditional approval that July, after a trial showed ~27% slowing of cognitive and functional decline over 18 months. For the first time, there was solid evidence that clearing amyloid could translate into meaningful clinical benefit, even if the effect was modest. A year later, in July 2024, the FDA approved donanemab (Kisunla). In the study, donanemab slowed decline by ~29–35%, with the largest benefits seen in people treated earlier in the disease course.
None of these drugs are cures, but they have shifted the field from no disease-modifying therapies at all to the first tangible, though modest, slowdowns in decline. The limited gains highlight two key issues. Timing: by the time symptoms appear, amyloid has been accumulating for over a decade, tau pathology is often entrenched, and neurodegeneration is already in motion. Starting treatment then may be too late to rescue neurons already lost. And target: most late-stage programs so far have aimed squarely at amyloid. Tau-directed antibodies are now advancing, with phase 3 trials ongoing, and it may be that targeting tau, or using combination therapies, will yield greater benefit.
Current diagnostics are used too late in disease progression
With Alzheimer’s, by the time symptoms become obvious, the disease has often been silently progressing for years and sometimes decades. Therefore, early detection will lead to a better prognosis. That’s where today’s practice collides with biology. Our healthcare system as it stands is built around reacting to problems rather than preventing them.
The Alzheimer’s Association Workgroup, composed of experts from the National Institute on Aging and the Alzheimer’s Association, has established guidelines to help clinicians diagnose Alzheimer’s. These guidelines emphasize shifting from a purely symptom-based approach to a biological definition of the disease. In this framework, Alzheimer’s is understood as a process that begins with measurable neuropathological changes, such as the accumulation of amyloid deposits, long before clinical symptoms appear. At present, however, presymptomatic testing is generally limited to individuals with a known genetic predisposition that puts them at higher risk and only in research settings.
Most people enter the system because of symptoms: a spouse or adult child notices that they repeat questions, don’t pay bills, or take wrong turns on a familiar drive. The clinician takes a careful history, screens cognition, checks mood and sleep, orders bloodwork to catch reversible contributors like thyroid disease or B-12 deficiency, and obtains structural imaging, such as MRI or CT, mainly to rule out other causes like stroke, tumors, or other injuries. These are appropriate first steps, but they are indirect for Alzheimer’s. They say little about amyloid and tau.
The most direct measure of Alzheimer’s is through Positron Emission Tomography. In PET imaging, a radioactive label is injected into the patient. These tracers are chemical compounds that specifically lodge themselves into Aβ sheets or tau tangles. By taking images with a camera, the physician can see the extent of plaque formation or tau spreading. There are currently three FDA-approved radiotracers for Aβ and one for tau. Most people have some low levels of plaques and tangles, but Alzheimer’s patients will show high levels of widespread distribution of these pathologies.
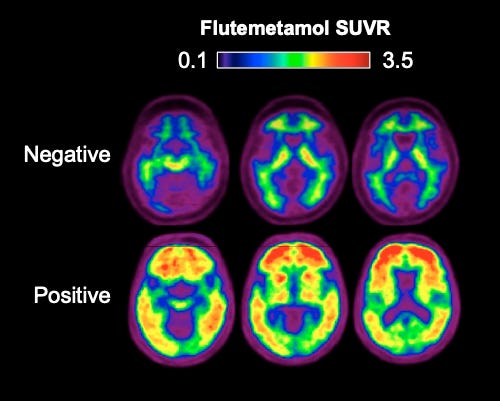
If PET imaging can be performed early, it has the potential to catch Aβ accumulation before the spread of tau pathology and neurodegeneration. However, PET scans, while accurate and sensitive, are expensive due to the cost of radiotracers. So, PET scans are only ordered when the physician is almost certain that the patient has Alzheimer’s.
A more recent form of testing is to perform a spinal tap. The fluid that surrounds the brain and spinal cord is called the cerebrospinal fluid (CSF). Small amounts of Aβ and tau naturally make their way into the CSF. By measuring these levels and comparing them to established healthy patterns, clinicians can detect the presence of Alzheimer’s-related changes with high accuracy. To get more specific, there are two major forms of amyloid-β: Aβ42 and Aβ40, both produced in the brain. Plaques deposited in the brain mostly consist of Aβ42. So, when plaques begin to form, the concentration of Aβ42 in the CSF falls, while Aβ40 remains relatively unchanged. Evidence from studies shows that the ratio of Aβ42/Aβ40 is consistently lower in Alzheimer’s patients than in healthy controls.
Similarly, tau pathologies can also be measured in the CSF. Tau is a normal protein that helps stabilize the internal scaffolding of neurons, but when it becomes abnormally phosphorylated (called p-tau) it tends to misfold and form tangles. Both total tau and p-tau circulate at low levels in the CSF, and their concentrations rise as tangles accumulate in the brain. Large studies and meta-analyses show that combining Aβ42/Aβ40 with p-tau makes CSF testing highly accurate, with over 90% sensitivity and specificity when compared against PET imaging or autopsy confirmation. In contrast, measuring Aβ alone achieves only around 80% sensitivity and specificity, meaning it produces more false positives and false negatives than the combined approach.
As with the PET scan, spinal taps are only performed when the patient has clear cognitive decline and other causes are ruled out. Performing the spinal tap requires a highly trained clinician and inserting a needle into the spinal canal. While any serious risks with the procedure are very low, people probably don’t want to do this unless necessary.
In real-world U.S. care, the accessibility of PET scans and CSF tests is still low. The rates vary between speciality clinics and primary care, but some data suggest a rate of 16% for speciality clinics and 10% in primary care settings. About 60-70% of patients receive structural imaging with CT or MRI. In short, the biological tests that could move diagnosis earlier and sharpen decisions were the least used.
We need a simple and accurate early diagnostic for Alzheimer’s
Blood tests, in contrast, are easy and common. That is why blood-based biomarkers have attracted so much attention. Small amounts of brain-derived Aβ and tau cross the blood-brain barrier, and with ultrasensitive assays, we can measure them. In May 2025, the FDA approved the first blood test to aid in diagnosing Alzheimer’s disease in adults 55 and older who are already being evaluated for cognitive impairment (which is a good start, but already late in the disease stage).
So how do these markers behave? On average, plasma Aβ42/40 is lower in people with amyloid plaques in the brain, but the change is modest, typically a 10–15% drop, compared to healthy controls. Notably, this 10-15% figure is in the advanced stage, and could be much lower in the early stages. Plasma p-tau levels, by contrast, show a much more robust increase with Alzheimer’s pathology and correlate strongly with both amyloid PET and tau PET scans. In large research cohorts, plasma p-tau can approach the diagnostic accuracy of CSF testing, often exceeding 90% sensitivity and specificity when combined with Aβ42/40.
But p-tau is a late signal. Phosphorylated tau reflects the stage when tangles are forming and spreading, which tends to occur closer to the onset of symptoms. If the goal is to diagnose Alzheimer’s in its earliest, pre-symptomatic phase, p-tau may not yet be elevated. In that scenario, we are left with Aβ alone, and Aβ by itself is not a strong predictor of disease progression. It flags the presence of pathology but not its severity, and its sensitivity and specificity are lower (around 80%) with more false positives and negatives compared to combined testing.
This means the current FDA-approved blood test is most powerful in people who already have signs of Alzheimer’s, not those in the silent phase we most want to reach. It is progress to have a blood test available at all, but the biology of the markers limits how early they can truly diagnose the disease. What we really need are biomarkers that change in lockstep with the very first shifts in Alzheimer’s biology, long before tau tangles and overt symptoms appear. Until then, blood tests will remain more of a tool for confirmation than for genuine early detection.
Where does this leave us?
Having accurate biomarkers for early diagnosis of Alzheimer’s, ones that are also simple and accessible, feels crucial. We are not there yet. In practice, only a small fraction of patients who present with memory concerns receive anything beyond neurological exams and basic imaging. PET scans and spinal taps remain limited by cost, availability, or invasiveness. Blood tests are promising, but they are still in their infancy, and the markers we have today are not perfect reflections of the earliest disease stages.
Why does this matter so much? Because diagnosis is not just about putting a label on a condition. An early and accurate diagnosis shapes care plans, allows patients and families to plan, and, perhaps most importantly, opens the door to intervening while the disease biology is still malleable. Alzheimer’s begins 15-20 years before symptoms, which means there is a long silent window where intervention could have the biggest impact. Without early diagnosis, we are always playing catch-up.
Of course, diagnostics are only one side of the equation. A test without an effective treatment is limited in value. The drugs we have so far offer only modest slowing of decline, and are used only after symptoms appear. Still, they show that disease modification is possible, and pairing them with earlier diagnosis may extend their benefit. That’s the hope of ongoing presymptomatic trials: to see if starting treatment before tau tangles and neurodegeneration set in can truly change the trajectory.
And then there’s the question of trust. The Alzheimer’s field has not been free of controversy. The high-profile fraud around a particular amyloid variant (Aβ*56) cast a long shadow and added to the public confusion. Fraud is bad, and ideally, we don’t want to be wasting resources and money. But it is also important to separate one fraudulent line of research from the thousands of independent studies that stand on solid ground. From what I understand, the overwhelming body of evidence on Aβ42, tau, and the biomarker cascade remains intact.
So, where does this leave us? We have a clearer map of how Alzheimer’s unfolds. We have our first disease-modifying therapies, even if modest. We have new blood tests that make biological diagnosis more accessible than before. None of these pieces is enough on its own. But together, they move us away from the old model of diagnosing late and treating symptomatically, and toward a future where Alzheimer’s can be detected earlier, targeted biologically, and perhaps one day, truly prevented.